再生医療製品の実用化が進む中、製造現場や品質保証の最前線にいらっしゃる皆様にとって、適切な製造管理体制の構築は避けて通れない重要な課題です。特に、従来の医薬品GMPと再生医療等製品固有のGCTP(Good Gene, Cellular, and Tissue-based Products Manufacturing Practice)の関連性や、法的な位置づけを正確に把握することは、査察対応のみならず、製品の安全性確保の根幹に関わります。
本記事では、製薬業界におけるGMP基準の概要を俯瞰しつつ、再生医療分野における適用のポイントや国際的な動向までを体系的に解説いたします。法令の背景にある「意図」を深く理解し、実務における判断の拠り所としていただけるよう、専門的な内容を噛み砕いてお伝えします。日々の業務における品質文化の醸成にお役立てください。
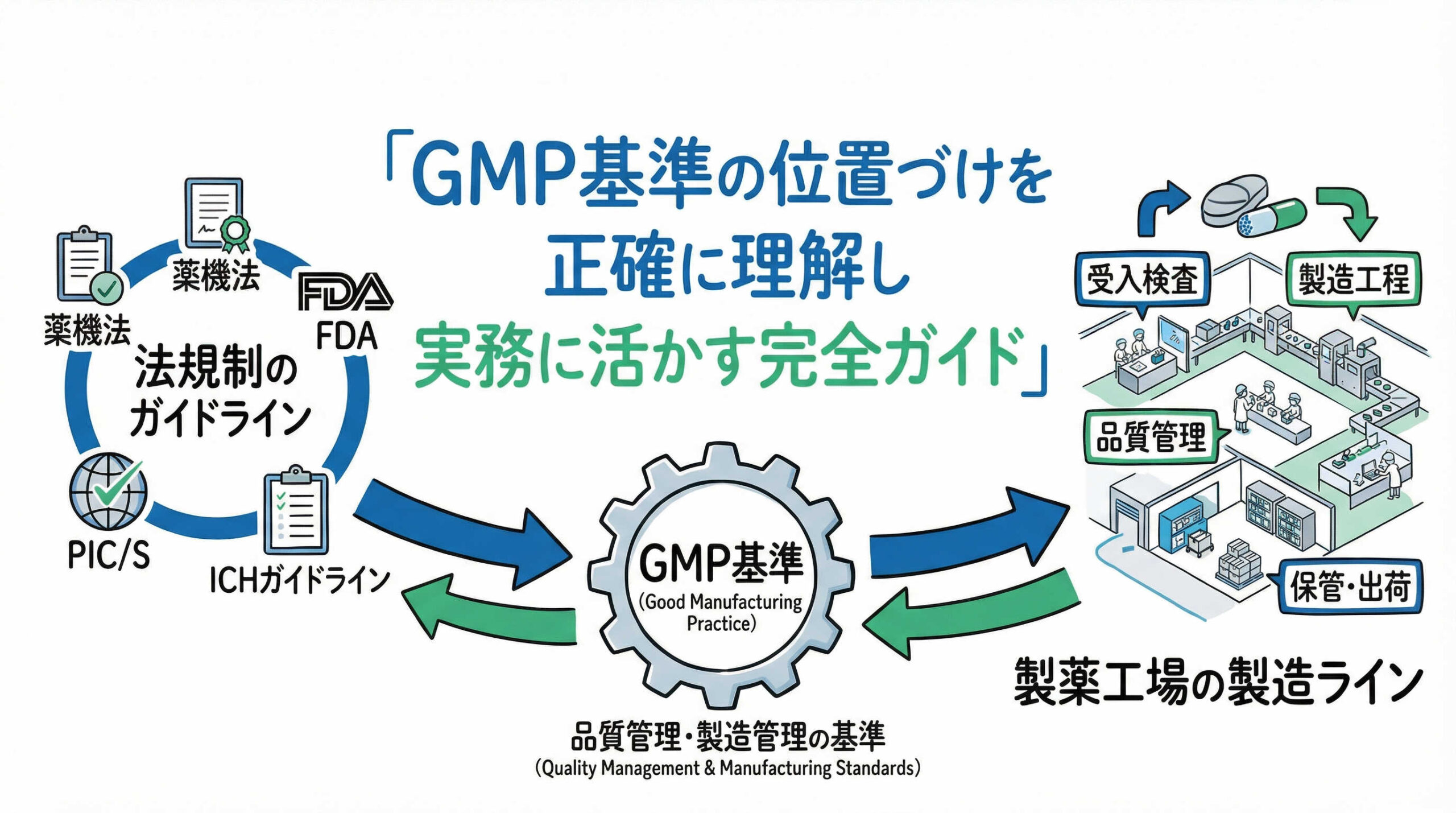
GMP基準の定義と製薬業界における法的位置づけ
医薬品や再生医療等製品の製造において、GMP(Good Manufacturing Practice)は単なるルールブックではなく、患者様の安全を守るための「砦」とも言える存在です。ここではまず、GMPの定義とその法的な位置づけについて、基本から整理してまいりましょう。
GMP(Good Manufacturing Practice)の基本概念と目的
GMPとは「医薬品の製造管理及び品質管理の基準」を指し、原料の入庫から製造、出荷に至るまでの全工程において、製品が安全に作られ、一定の品質が保たれるようにするための要件を定めたものです。
その目的は、科学的な根拠に基づいた管理手法を用いることで、不良品の発生を防ぎ、高品質な医薬品を安定して供給することにあります。製造現場においては、誰が作業しても同じ品質の結果が得られるよう、標準化された手順の遵守が求められます。これは、最終製品の検査だけでは発見できないリスクを製造プロセスの中で低減させるという、品質保証の核心部分を担っています。
薬機法におけるGMP省令の法的根拠と強制力
日本においてGMPは、いわゆる「薬機法(医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律)」に基づき、厚生労働省令として定められています。これは任意のガイドラインではなく、法的拘束力を持つ「省令」である点が極めて重要です。
製造販売業の許可要件の一つとしてGMP適合が含まれており、これを遵守しなければ製品を市場に出すことはできません。違反した場合には、業務停止命令や許可の取り消しといった行政処分の対象となります。したがって、企業にとってはコンプライアンスの最優先事項であり、経営基盤そのものを支える法的義務として位置づけられています。
製薬バリューチェーン全体におけるGMPの役割
製薬産業のバリューチェーンにおいて、GMPは研究開発から商用生産、そして市販後の製品ライフサイクル全体に関わる重要な役割を果たしています。開発段階で確立された品質設計(Quality by Design)を、実際の製造現場で具現化し、維持・管理するのがGMPの実務です。
また、GQP(品質管理の基準)やGVP(安全管理の基準)とも密接に連携し、製造所からの品質情報のフィードバックや、市場での不具合発生時の原因究明においても、GMPに基づく製造記録が不可欠な証拠となります。つまりGMPは、サプライチェーン全体における品質の信頼性を担保する共通言語としての機能を果たしているのです。
再生医療等製品におけるGCTPとの関係性と法的区分
再生医療等製品に関しては、従来の医薬品GMPとは別に「GCTP省令」が適用されます。これは、細胞や組織といった原材料の特殊性を考慮し、医薬品GMPの考え方をベースにしつつも、再生医療等製品特有の要件を盛り込んだ基準です。
法的には、医薬品GMPと同様に薬機法に基づく省令として位置づけられていますが、対象となる製品の特性(不均一性や個体差など)に応じた柔軟な運用が認められている点が特徴です。実務担当者としては、自社製品がどちらのカテゴリーに属するかを明確にし、それぞれの省令が求める要求事項の微妙な差異(例えば、バリデーションとベリフィケーションの使い分けなど)を正しく理解しておく必要があります。
GMPの3原則と品質保証システムが重要視される背景
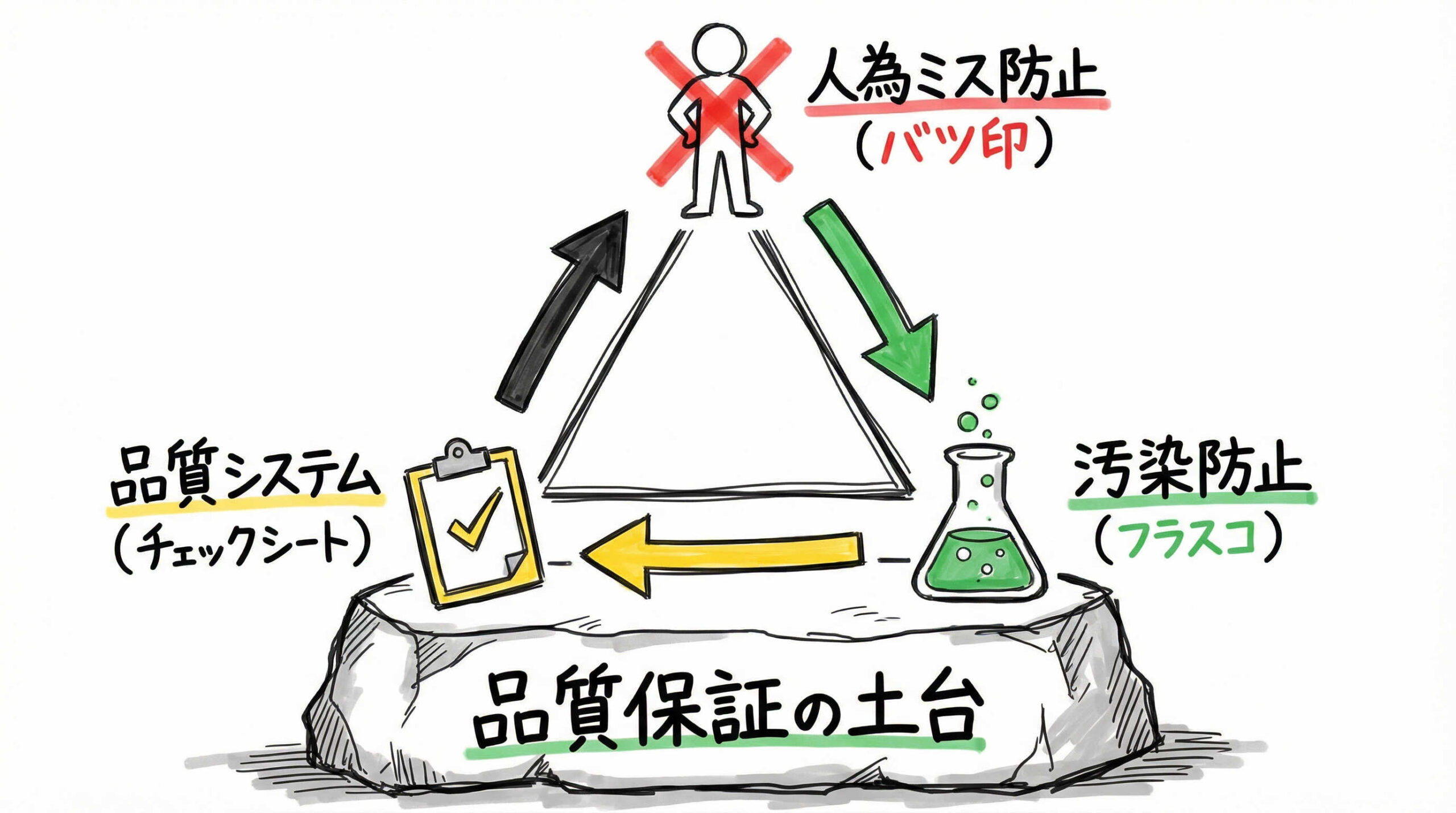
GMPがなぜこれほどまでに厳格に運用されるのか、その根底には「GMPの3原則」と呼ばれる基本的な考え方があります。これらは品質保証システムを構築する上での羅針盤となるものです。それぞれの原則が持つ意味を再確認しましょう。
人為的な誤りを最小限に抑えるための仕組み
第一の原則は「人為的な誤りを最小限にすること」です。人間が作業を行う以上、ミスを完全にゼロにすることは困難ですが、システムや手順によって限りなくゼロに近づけることは可能です。
具体的には、以下のような対策が講じられます。
- 二重チェック体制: 重要な工程や計算において、ダブルチェックを行う
- 明確な手順書(SOP): 曖昧さを排除した具体的な作業指示
- 誤認防止の表示: 原料や製品のステータスを明確にするラベル管理
これらは、作業者の記憶や注意のみに頼るのではなく、仕組みとしてミスを防ぐための重要なアプローチです。
医薬品の汚染および品質低下を防止する対策
第二の原則は「医薬品の汚染および品質低下を防止すること」です。医薬品や再生医療等製品は、微量な異物混入や微生物汚染が患者様の生命に関わる重大な結果を招く可能性があります。
そのため、製造環境の清浄度管理(クリーンルームの運用)や、交差汚染(クロスコンタミネーション)を防ぐための動線分離、設備洗浄の徹底が求められます。特に再生医療分野では、無菌操作が前提となる工程が多く、ハードウェア(設備)とソフトウェア(衛生管理手順)の両面から、汚染リスクを徹底的に排除する対策が不可欠です。
高い品質を恒常的に保証するシステムの設計
第三の原則は「高い品質を恒常的に保証するシステムを設計すること」です。一度だけ高品質な製品ができれば良いのではなく、いつ製造しても同じ品質を再現できなければなりません。
これを実現するために、製造設備の保守点検や計測機器の校正(キャリブレーション)を定期的に行い、プロセスの状態が常に管理された範囲内にあることを監視します。また、変更管理や逸脱管理といった品質システム(PQS)を適切に運用し、品質に影響を与える要因をコントロールし続ける体制こそが、恒常的な品質保証の基盤となります。
製品試験だけでなく製造プロセス全体で品質を作り込む意義
これら3原則を貫く理念として、「品質は試験だけでなく、製造プロセス全体で作り込む」という考え方があります。最終製品の抜き取り検査だけでは、全ての製品の品質を保証するには限界があるからです。
原材料の受け入れから、製造工程の一つひとつにおいて定められた基準を満たしていくことで、結果として最終製品の品質が担保されるというアプローチです。これは現代のGMP/GCTPにおいて最も重視される概念であり、プロセスバリデーションや継続的な工程確認(Process Verification)の重要性が叫ばれる理由でもあります。プロセス全体への深い理解が、真の品質保証につながるのです。
医薬品GMPと再生医療等製品GCTPの相違点と適用実務
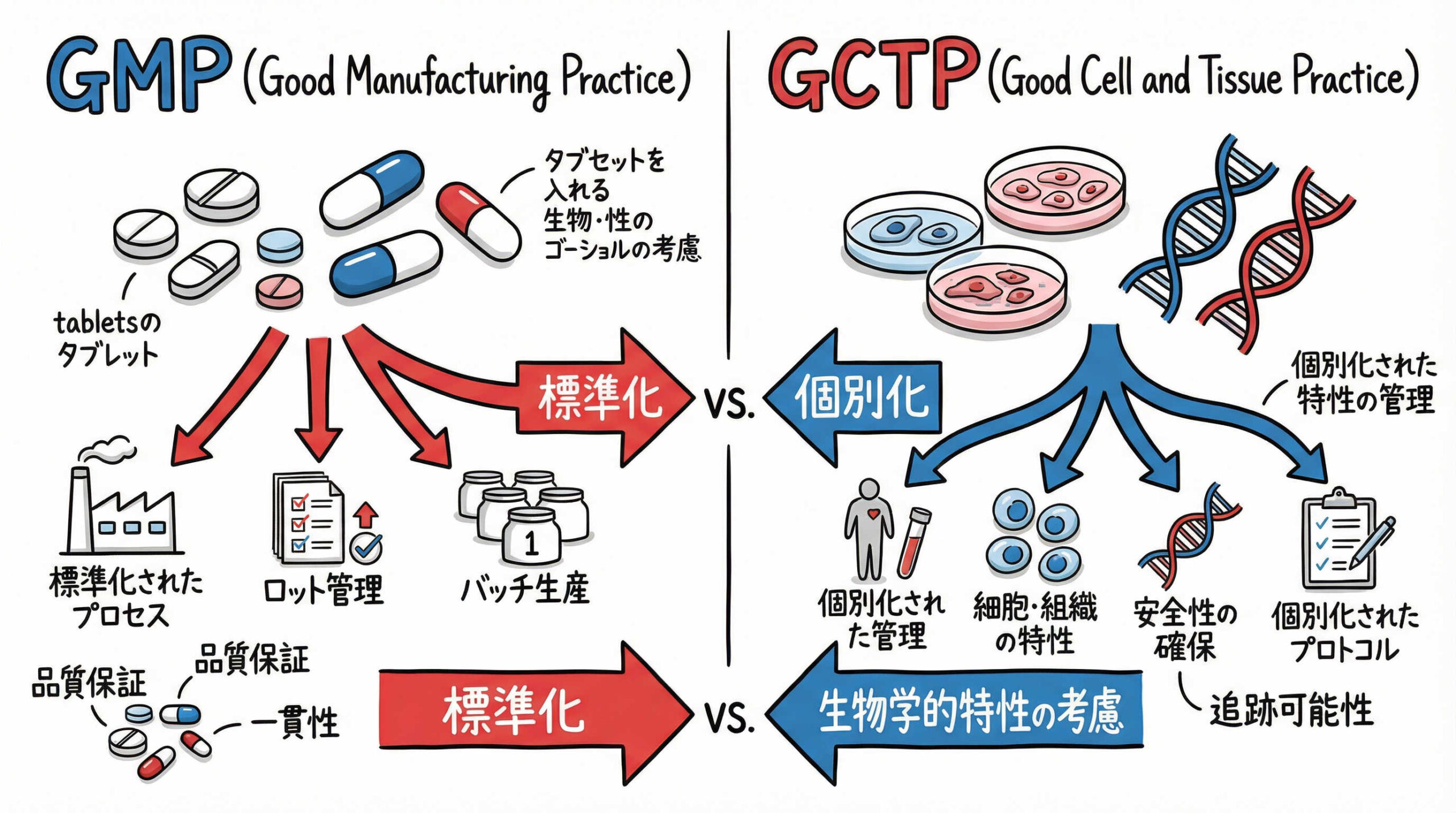
再生医療に携わる皆様にとって、従来の医薬品GMPとGCTPの違いを理解することは実務上極めて重要です。ここでは、両者の適用範囲や管理手法の相違点に焦点を当て、現場での適用実務について解説します。
従来の医薬品GMP省令が適用される範囲と特徴
従来の医薬品GMP省令は、主に低分子医薬品やバイオ医薬品などを対象としています。これらの製品は、化学合成や培養タンクでの大量生産が可能であり、ロットサイズが大きく、均質な製品を製造することが前提となっています。
そのため、製造プロセスは高度に自動化・標準化されやすく、統計的な手法を用いた品質管理が馴染みやすい特徴があります。3ロットの一貫性を示すプロセスバリデーションが厳格に求められるのも、製品の均質性が担保できるという前提があるからです。大量生産品におけるリスク管理の集大成とも言えるでしょう。
再生医療等製品GCTP省令における特異性と柔軟性
一方、再生医療等製品GCTP省令は、ヒトの細胞や組織を用いる製品を対象としており、その特性に応じた特異性と柔軟性が認められています。特に自家移植(患者様自身の細胞を用いる場合)のような極めて小規模なロットや、個体差による原材料のばらつきが避けられないケースが多々あります。
GCTPでは、こうした事情を考慮し、画一的な基準を適用するのではなく、製品ごとの特性に基づいたリスクベースのアプローチが重視されます。無菌操作の重要性が極めて高い一方で、科学的な妥当性があれば、運用面での柔軟な対応が可能となる余地が残されているのです。
原材料の生物由来性に起因する品質管理の難易度
GCTP運用における最大の課題の一つは、原材料の生物由来性に起因する品質管理の難易度です。ドナーの状態や採取部位によって細胞の性質が変動するため、出発原料の均質性を完全に保証することは容易ではありません。
このため、受入試験の基準設定や、製造工程内での管理幅の設定には高度な専門知識と経験が求められます。単に規格内であれば良しとするだけでなく、原材料の変動が最終製品の品質にどのような影響を与えるかを理解し、製造プロセス全体でその変動を吸収できるような管理戦略(コントロール戦略)を構築することが、品質保証担当者の腕の見せ所と言えるでしょう。
バリデーションとベリフィケーションの概念と使い分け
医薬品GMPでは、あらかじめ設定した条件下で期待通りの結果が得られることを検証する「バリデーション」が必須です。しかし、再生医療等製品、特に自家細胞製品などでは、検体数が限られるため、統計的に有意なバリデーションを実施することが困難な場合があります。
そこでGCTPでは、製造ごとの結果を確認する「ベリフィケーション」という概念が重要視されます。これは、プロセス全体を固定的に検証するだけでなく、個々の製造ロットごとに記録やデータを照査し、品質が確保されているかを確認する手法です。実務においては、製品特性に応じてバリデーションとベリフィケーションを適切に使い分ける、あるいは組み合わせる判断が求められます。
国際的なGMP基準の動向と日本国内規制への影響
製薬業界はグローバル化が進んでおり、国内規制だけでなく国際的な基準への対応も不可欠です。ここでは、世界のGMP動向が日本の規制や皆様の業務にどのような影響を与えているかを見ていきましょう。
PIC/S(医薬品査察協定・医薬品査察共同スキーム)への加盟と整合性
日本は2014年にPIC/S(医薬品査察協定・医薬品査察共同スキーム)に加盟しました。これは各国の査察当局間の国際的な枠組みであり、GMP基準の整合性を図ることを目的としています。
PIC/Sへの加盟により、日本のGMP省令も国際標準であるPIC/S GMPガイドラインとの整合性が強化されました。これにより、国内の製造所であっても、実質的には世界標準レベルの品質管理体制が求められるようになっています。グローバルな視点を持つことは、もはや輸出企業だけでなく、国内供給を行う企業にとっても標準的な要件となりつつあるのです。
ICHガイドライン(Q8, Q9, Q10)に基づく医薬品品質システム(PQS)
日米欧の規制当局と製薬業界で構成されるICH(医薬品規制調和国際会議)では、品質に関するガイドライン(Qトリオ)が策定されています。
- ICH Q8: 製剤開発(Quality by Designの導入)
- ICH Q9: 品質リスクマネジメント
- ICH Q10: 医薬品品質システム
これらは現代のGMP運用の根幹をなす概念です。単に手順を守るだけでなく、科学的知見に基づいた開発、リスクに応じた管理、そして継続的な改善サイクル(PQS)を回すことが求められます。日本のGMP/GCTP省令もこれらの概念を取り入れて改正されており、現場での運用にも深く浸透しています。
米国cGMPやEU-GMPなど海外規制要件との調和
海外展開を見据える場合、米国のcGMP(current GMP)や欧州のEU-GMPへの対応は避けて通れません。これらの規制は日本の基準と大枠では共通していますが、細部の要求事項や査察の視点に違いがあります。
例えば、米国FDAはデータインテグリティ(データの完全性)や無菌保証に対して非常に厳しい視点を持っています。また、EUでは適格性評価者(QP)の役割が明確に定義されています。国内規制への適合をベースにしつつ、これら海外規制とのギャップ分析を行い、より高いレベルでの調和を図ることは、企業の競争力を高めることにも繋がります。
相互承認協定(MRA)による輸出入時のメリット
国際的な基準整合が進むことの大きなメリットとして、相互承認協定(MRA)の活用が挙げられます。これは、相手国の査察結果を自国のものとして受け入れる仕組みです。
日本と欧州間などでMRAが適用されると、輸出入時の重複した試験検査や査察が免除または軽減されるため、迅速な製品供給やコスト削減が可能となります。再生医療等製品においても、将来的に国際的な調和が進めば、こうした枠組みの恩恵を受けられる可能性が広がります。国際基準を意識した体制構築は、将来のビジネスチャンスを広げる投資とも言えるでしょう。
GMP/GCTP適合性調査への対応と体制構築の具体策

GMPやGCTPの要件を満たしているかを確認される適合性調査は、製造販売承認の要です。ここでは、調査への具体的な対応策や、日頃から構築しておくべき体制について、実務的な視点から掘り下げます。
製造販売承認申請時における適合性調査のプロセス
新しい医薬品や再生医療等製品の製造販売承認申請を行う際、PMDA(医薬品医療機器総合機構)や都道府県による適合性調査が実施されます。これは、申請資料(CTD)に記載された通りに製造管理・品質管理が行われているかを現場で確認するプロセスです。
申請から調査実施までの期間には、模擬査察(モック査察)を行うなどして、指摘事項となり得る不備を事前に洗い出すことが重要です。特にGCTPでは、開発段階からの変更管理の履歴や、同等性評価のデータが厳しくチェックされるため、申請資料と現場の実態に乖離がないよう、綿密な整合性確認が求められます。
定期的な実地調査および書面調査への準備対応
適合性調査は初回だけでなく、製造販売承認取得後も定期的(原則5年ごと)に実施されます。これを更新調査や定期適合性調査と呼びます。ここでは、前回の調査以降の変更管理状況や、逸脱・苦情への対応履歴が重点的に確認されます。
また、実地調査だけでなく、書面調査が行われる場合もあります。日頃から製造記録や品質関連文書を整理し、求められた資料を即座に提示できる状態(Inspection Readiness)を維持しておくことが、スムーズな調査対応の鍵となります。日常業務そのものが査察準備であるという意識を持つことが大切です。
構造設備(ハードウェア)の要件適合と維持管理
ハードウェアである構造設備の適合性は、GMP/GCTPの基本要件です。空調システム(HVAC)や製造用水システム、製造機器などが適切に設計・据え付けられ、その性能が維持されていることを証明しなければなりません。
具体的には、適格性評価(DQ, IQ, OQ, PQ)の記録や、定期的なメンテナンス、キャリブレーションの実施記録が重要です。再生医療では特に、細胞調製施設の無菌性保証(清浄度区分や室圧制御)がクリティカルな要素となるため、モニタリングデータのトレンド分析を含めた徹底的な維持管理が求められます。
手順書(SOP)および製造記録等の文書管理(ソフトウェア)の徹底
ソフトウェアの側面である文書管理は、GMP活動の証拠そのものです。「記録がないことは実施していないことと同じ」と見なされる世界において、SOP(標準作業手順書)の整備と、それに基づく正確な製造記録の作成は必須です。
手順書は、作業者が迷いなく実行できる具体性を持ち、かつ最新版管理が徹底されている必要があります。また、記録の訂正ルール(見え消し修正など)や承認フローの遵守も重要です。文書体系全体が論理的に整合しており、トレーサビリティ(追跡可能性)が確保されているか、定期的に自己点検を行うことをお勧めします。
データインテグリティ(DI)確保に向けた教育訓練
近年、規制当局が最も注視しているのがデータインテグリティ(DI:データの完全性)です。これは、データが正確で、完全で、信頼できる状態であることを指します。電子データの改ざん防止機能や、アクセス権限の管理はもちろん、紙記録においてもALCOA+原則(帰属性、判読性、同時性、原本性、正確性など)の遵守が求められます。
システム的な対策に加え、重要なのは「不正を行わせない、隠蔽させない」組織風土づくりと教育訓練です。DIの重要性を全従業員に浸透させ、正直な報告が評価される文化を醸成することが、コンプライアンス違反を防ぐ最後の防波堤となります。
まとめ

本記事では、GMP基準の概要から、製薬業界における法的位置づけ、そして再生医療等製品GCTPとの関連性について解説してまいりました。
GMP/GCTPは、単なる規制への対応ではなく、患者様に安全で高品質な製品を届けるための「品質文化」そのものです。ハード・ソフト両面からの管理体制構築、国際基準との調和、そしてデータインテグリティの確保は、一朝一夕に成し遂げられるものではありません。しかし、日々の業務における誠実な積み重ねこそが、確固たる信頼を築く唯一の道です。
再生医療という先端分野において、皆様が構築される品質保証体制が、多くの患者様の希望となることを願っております。今回の内容が、実務における判断の一助となれば幸いです。
GMP基準の概要と製薬業界での位置づけについてよくある質問

GMP基準の概要と製薬業界での位置づけに関して、よくお問い合わせいただく質問をまとめました。実務の参考としてご活用ください。
-
Q1. GMPとGQPの違いは何ですか?
- A1. GMP(Good Manufacturing Practice)は「製造所」における製造管理・品質管理の基準であり、製品を作る現場が対象です。一方、GQP(Good Quality Practice)は「製造販売業者」における品質管理の基準であり、市場への出荷判定や適正な品質情報の収集・分析など、製品全体の品質保証責任を担う機能が対象となります。両者は車の両輪のように連携して機能します。
-
Q2. 再生医療等製品でも医薬品GMPのガイドラインを参考にして良いですか?
- A2. はい、基本的には参考になります。GCTPは医薬品GMPの考え方をベースにしているため、無菌操作や文書管理、教育訓練などの基本原則は共通しています。ただし、バリデーションの手法や原材料管理など、再生医療特有の柔軟性が求められる部分については、GCTP省令や関連する通知・ガイドラインを優先して参照する必要があります。
-
Q3. 治験薬製造段階でもGMPへの適合は必須ですか?
- A3. はい、必須です。「治験薬GMP」という基準に従う必要があります。商用生産のGMPと比較して一部柔軟な運用が認められていますが、被験者の安全確保と治験データの信頼性確保の観点から、基本的な品質保証体制は確立されていなければなりません。将来の承認申請を見据え、開発段階からGMP体制を整えておくことが推奨されます。
-
Q4. データインテグリティ(DI)対応で最初にすべきことは何ですか?
- A4. まずは現状のリスク評価(ギャップ分析)を行うことをお勧めします。使用している機器やシステムがDI要件(監査証跡機能やセキュリティ設定など)を満たしているかを確認し、不備がある場合は暫定的な運用対応策を策定します。同時に、全従業員に対してDIの重要性を伝える教育訓練を実施し、意識改革を進めることが重要です。
-
Q5. 海外への輸出を考えていない場合、PIC/S GMPを意識する必要はありますか?
- A5. はい、意識する必要があります。日本のGMP省令自体がPIC/S GMPガイドラインと整合するように改正されているため、国内向け製品であっても、実質的にはPIC/Sレベルの管理が求められます。また、将来的にビジネス環境が変化する可能性もあるため、国際基準に準拠した体制を構築しておくことは、企業としてのリスク管理や競争力強化につながります。