近年、再生医療等製品の実用化が急速に進展するなかで、その製造管理および品質管理の基準である「GCTP」への理解と対応が急務となっています。従来の医薬品に適用される「GMP」とは異なり、生きた細胞や組織を扱う再生医療では、特有の品質リスクや製造上の課題が存在するためです。
これから再生医療等製品の製造体制を構築しようとする製薬企業やバイオベンチャーの担当者様にとって、両省令の違いを明確に把握することは、コンプライアンス遵守のみならず、製品の安全性と有効性を担保する上で極めて重要です。本記事では、GMPとGCTPの定義や関係性、そして実務における具体的な要求事項の差異について、専門的な視点から分かりやすく解説します。再生医療特有の品質基準を深く理解し、適切な管理体制の構築にお役立てください。
GMPとGCTPの違いと関係性:再生医療における位置づけ
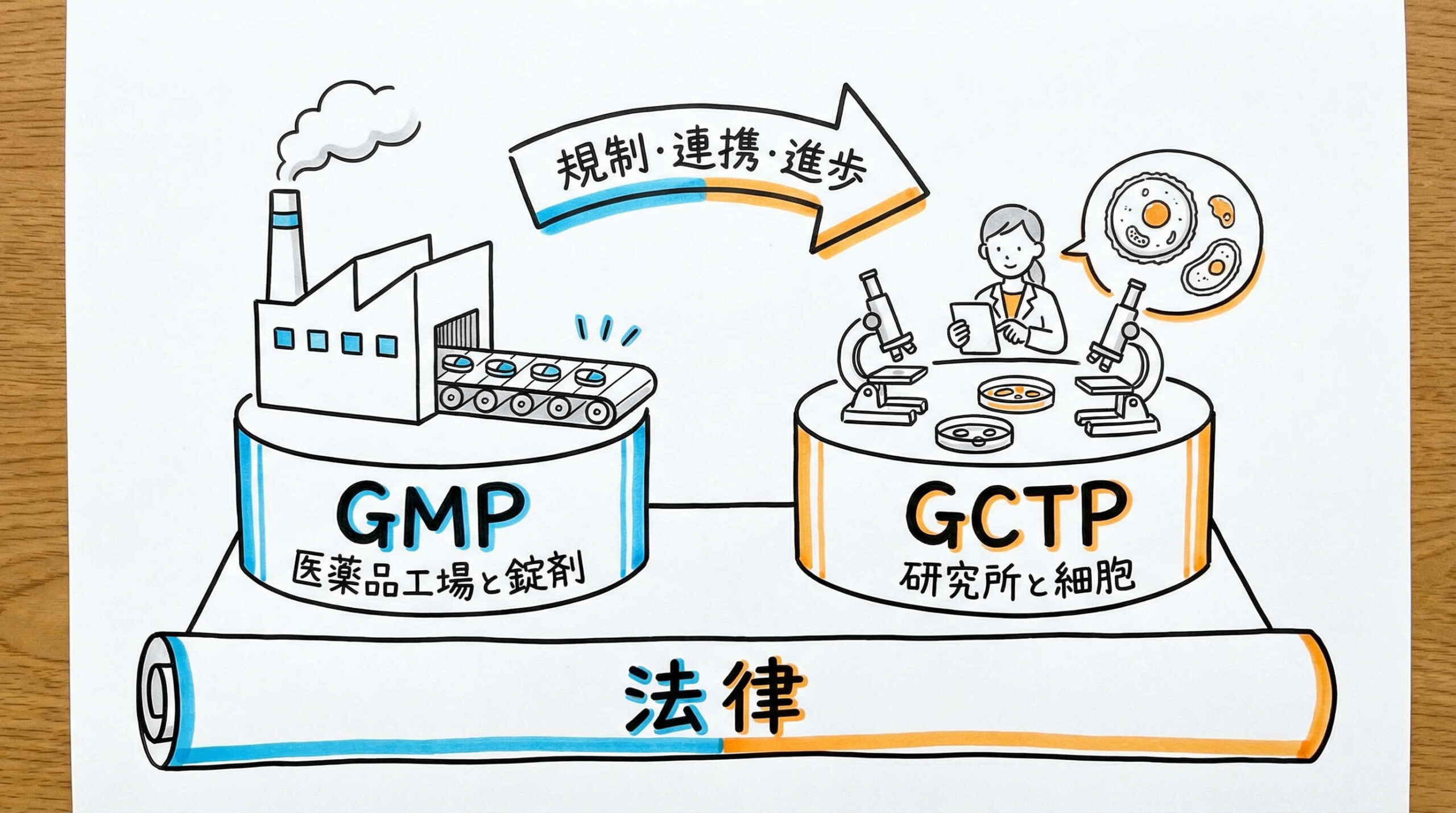
まずは、医薬品製造の基本となるGMPと、再生医療等製品に特化したGCTPの定義、そして両者の法的な位置づけについて整理しましょう。これらは単なるルールの違いではなく、取り扱う製品の特性に根ざした必然的な差異に基づいています。ここでは、それぞれの定義と薬機法における相互関係について解説します。
医薬品製造におけるGMP(Good Manufacturing Practice)の定義
GMP(Good Manufacturing Practice)は、「医薬品及び医薬部外品の製造管理及び品質管理の基準」に関する省令です。その目的は、製造所における人為的な誤りを最小限にし、汚染や品質低下を防止し、高度な品質保証システムを確立することにあります。
主に低分子医薬品やバイオ医薬品を対象としており、均質な製品を恒常的に大量生産することを前提としています。原材料の受入から製造、出荷に至るまでの全工程において、科学的な妥当性に基づいた厳格な管理が求められるのが特徴です。医薬品製造においては、このGMPへの適合が製造業許可の要件となります。
再生医療等製品におけるGCTP(Good Gene, Cellular, and Tissue-based Products Manufacturing Practice)の定義
GCTP(Good Gene, Cellular, and Tissue-based Products Manufacturing Practice)は、「再生医療等製品の製造管理及び品質管理の基準」に関する省令です。2014年の薬機法改正に伴い、再生医療等製品という新たなカテゴリーが新設されたことに合わせて制定されました。
GCTPは、人や動物の細胞・組織を加工して製造される製品を対象としています。GMPの基本概念を踏襲しつつも、原材料の不均一性や無菌操作の重要性など、再生医療等製品特有の事情を考慮した柔軟かつ厳格な基準が設けられています。これにより、画一的な管理が難しい細胞製品の品質を担保します。
医薬品医療機器等法(薬機法)における両省令の適用範囲と相互関係
医薬品医療機器等法(薬機法)において、GMP省令とGCTP省令は兄弟のような関係にありますが、適用される製品カテゴリーが明確に区分されています。従来の医薬品にはGMPが適用される一方、再生医療等製品にはGCTPが適用されます。
GCTPは、GMPの構成をベースにしつつ、再生医療特有の要件を追加・変更したものです。例えば、GMPでは「製品標準書」を作成しますが、GCTPでは「再生医療等製品製品標準書」となり、記載事項にも差異があります。両省令は相互に矛盾するものではなく、製品特性に応じて最適化された基準であると理解することが、適切な法規制対応の第一歩です。
なぜ再生医療にはGCTPが必要なのか:生物由来製品の特性
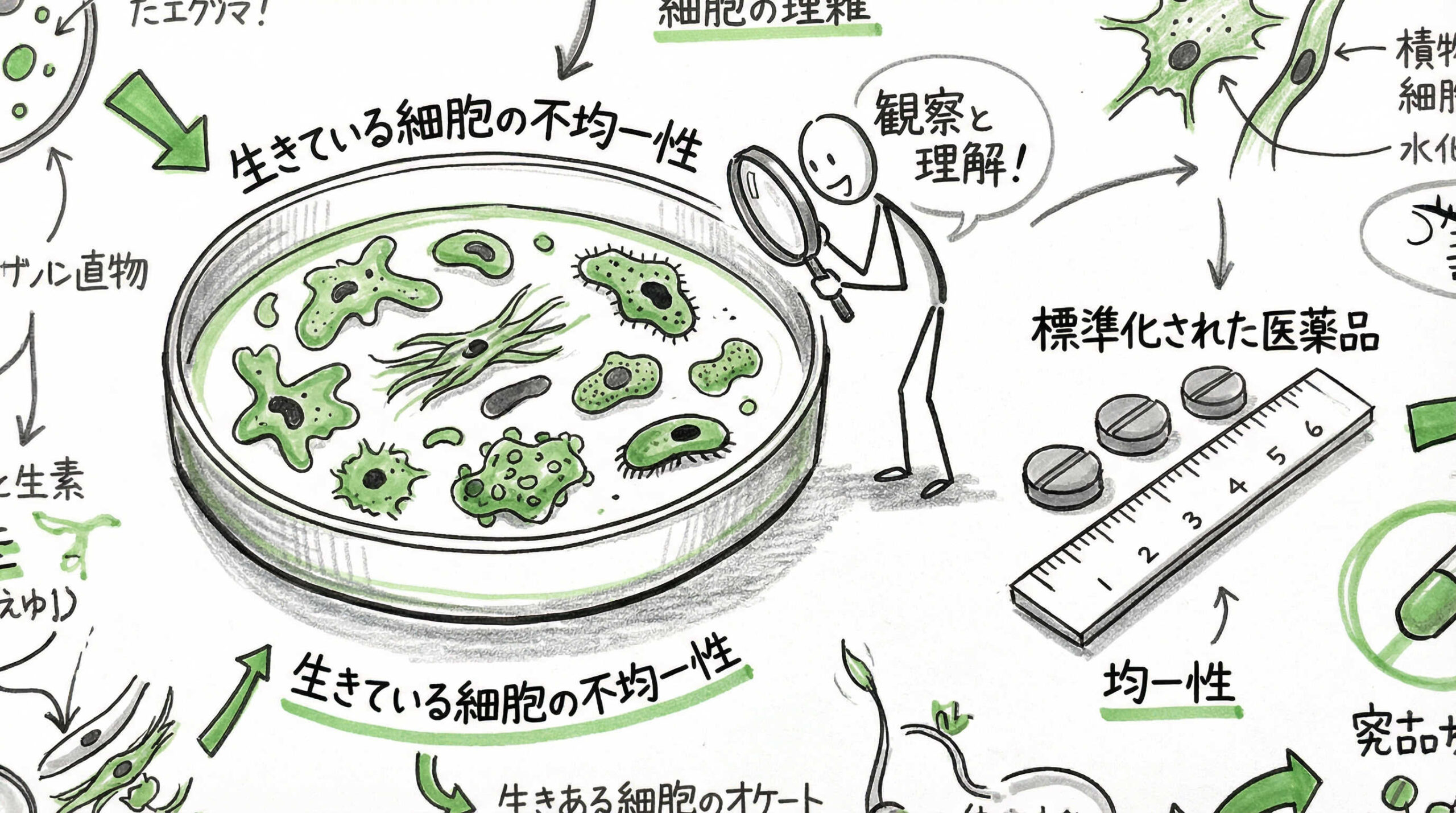
なぜ従来のGMPをそのまま適用せず、わざわざGCTPという新たな基準を設ける必要があったのでしょうか。その理由は、再生医療等製品が「生きている細胞」を扱う生物由来製品であるという点に尽きます。ここでは、GCTPが必要とされる背景にある、再生医療特有の3つの特性について詳しく見ていきましょう。
原材料となる細胞・組織の不均一性と個体差への対応
再生医療等製品の最大の特徴は、原材料となる細胞や組織がドナー(提供者)に由来するため、どうしても個体差が生じることです。化学合成される医薬品とは異なり、出発原料が不均一であるため、最終製品の品質を一定に保つことが非常に困難です。
GCTPでは、この「不均一性」を前提とした管理が求められます。原材料の受入試験を厳格に行うだけでなく、製造プロセス全体を通じて変動をモニタリングし、許容範囲内に収めるための高度なプロセス管理が必要となります。個体差を考慮した柔軟な製造条件の設定が認められる場合があるのも、GCTPならではの特徴でしょう。
最終製品での無菌化(ターミナルステリライゼーション)の困難性
一般的な医薬品、特に注射剤などでは、最終製品の段階で加熱滅菌やガス滅菌を行う「ターミナルステリライゼーション」が推奨されます。しかし、再生医療等製品は生きた細胞そのものが薬効を発揮するため、最終工程での滅菌処理を行うことができません。
そのため、製造工程の最初から最後まで、無菌性を維持したまま製造する「アセプティックプロセッシング(無菌操作法)」が絶対条件となります。GCTPでは、GMP以上に無菌操作区域の管理や、作業員の無菌操作手技に対する要求が厳しく設定されており、微生物汚染のリスクを極限まで低減する設計が求められます。
製品の有効期間の短さと全数検査の限界
多くの再生医療等製品、特に自家細胞を用いた製品では、有効期間が極めて短い場合があります。製品によっては、製造後数時間から数日以内に患者へ投与しなければならないケースも少なくありません。
これにより、従来のGMPで求められるような、長期間を要する無菌試験の結果を待ってから出荷するという運用が困難になります。GCTPでは、こうした特性に対応するため、製造工程中のモニタリングデータや速報的な試験結果に基づいて出荷を判定する仕組みや、全数検査が不可能な場合の代替措置など、製品特性に応じた品質保証のアプローチが許容されています。
GMPとGCTPの具体的な要求事項の比較
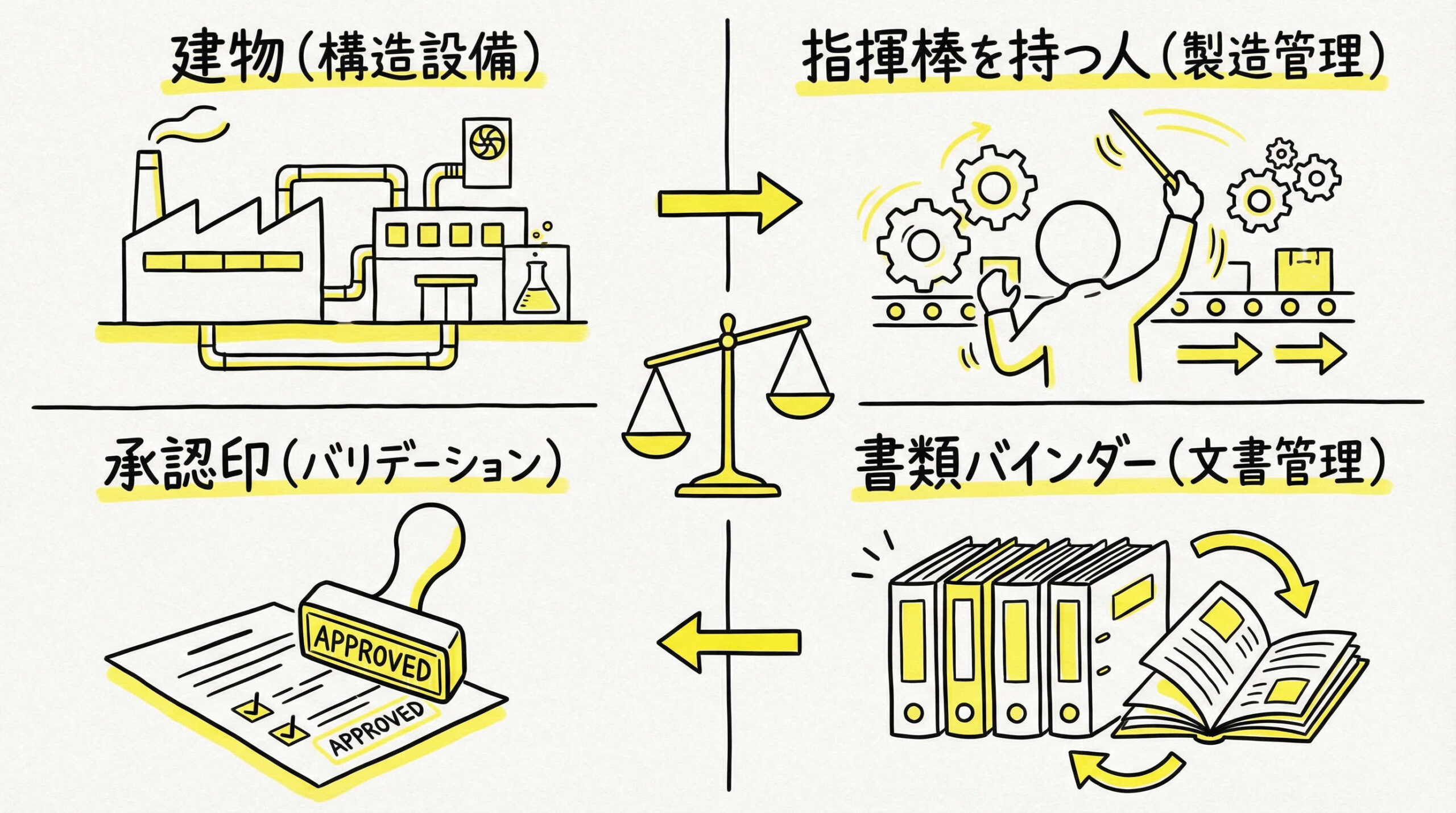
概念的な違いを理解したところで、次は実務レベルでの具体的な要求事項の違いに焦点を当ててみましょう。構造設備、製造管理、バリデーション、文書管理の4つの視点から、GMPとGCTPの運用の違いを比較します。これらの差異を正確に把握することが、現場での適切なSOP(標準作業手順書)作成につながります。
構造設備(ハードウェア):無菌操作区域と汚染防止対策
構造設備(ハードウェア)に関しては、両者ともに汚染防止が最優先ですが、GCTPでは特に「交差汚染の防止」と「無菌性の確保」が厳格です。細胞調製施設(CPF)では、清浄度区分(グレードA~D)の明確な管理が求められます。
特に、複数の患者の細胞を同一施設で扱う場合、検体の取り違えやウイルス等の交差汚染を防ぐため、作業区域の専用化や、時間的・空間的な分離、空調システムによる差圧管理が重要になります。また、安全キャビネットやアイソレータなどの閉鎖系システムの活用が、GMP以上に強く推奨される傾向にあります。
製造管理(ソフトウェア):プロセス管理の重要性と手法
製造管理(ソフトウェア)の面では、プロセス管理(工程管理)の重要性がGCTPにおいてより高まります。原材料の不均一性をカバーし、一定の品質を確保するためには、最終製品の試験だけでなく、製造プロセスの一つひとつが適切に実施されたかどうかの記録が品質保証の鍵となるからです。
具体的には、細胞の増殖率、形態、生存率などを工程ごとに細かく確認し、規格から外れた場合の処置手順をあらかじめ定めておく必要があります。GMPと比較して、製造工程そのものが品質を作り込むという「Quality by Design」の考え方が、より現場運用に直結していると言えるでしょう。
バリデーションとベリフィケーションの概念上の相違点
バリデーション(妥当性確認)はGMPの基本ですが、GCTPでは「ベリフィケーション(検証)」という概念が頻繁に登場します。これは、原材料の個体差が大きいため、3ロットの製造で恒常性を証明する従来のプロセスバリデーションだけでは不十分な場合があるからです。
そのため、GCTPでは、製造ごとの結果が判定基準に適合しているかを都度確認するベリフィケーションを併用するアプローチが取られます。画一的なバリデーションだけでなく、個々の製造ロットに対する検証を重視することで、生物由来製品特有の変動リスクを管理します。
文書管理と記録の保存期間に関する規定の違い
文書管理については、基本的な体系は似ていますが、記録の保存期間に大きな違いがあります。再生医療等製品は、体内に投与された後、長期間にわたってその影響が持続する可能性があるためです。
特に、人の血液や細胞を原料とする「特定生物由来製品」に該当する場合、その記録は使用後少なくとも30年間(通常の再生医療等製品でも10年以上が一般的)という極めて長期の保存が義務付けられています。これに伴い、紙媒体だけでなく電子データとしての保存や、長期的な検索性を確保したアーカイブ体制の構築が、GMP以上に重要な課題となります。
GCTP省令準拠における実務上の重要管理ポイント

GCTP省令に準拠した製造体制を構築・運用する際、現場担当者が特に注力すべき管理ポイントがいくつか存在します。これらは査察においても重点的に確認される項目であり、製品の品質と安全性を左右する要石です。実務において優先的に取り組むべき5つの重要ポイントを解説します。
交差汚染(クロスコンタミネーション)防止のための厳格な管理体制
再生医療の製造現場において、最も恐れるべきリスクの一つが交差汚染(クロスコンタミネーション)です。他人の細胞や異なるウイルスが混入することは、患者にとって致命的な結果を招きかねません。
これを防ぐため、同一エリアで複数の製品を扱う場合は、作業時間をずらす「時間的分離」や、使用する機器や器具を専用化するなどの対策を徹底する必要があります。また、清掃手順のバリデーション(クリーニングバリデーション)を行い、前の作業の痕跡が完全に除去されていることを科学的に証明することも不可欠です。一方通行の動線確保も有効な手段となります。
ドナーから患者までの双方向トレーサビリティの確保
GCTPでは、ドナーから採取された細胞がどの製品に使用され、最終的にどの患者に投与されたか、また逆に、製品からドナーを特定できる「双方向のトレーサビリティ」の確保が義務付けられています。
これは、感染症等の副作用が発生した際に、迅速に原因を究明し、被害拡大を防ぐためです。製造記録、試験記録、配送記録を紐づけて管理するシステムが必要となりますが、同時にドナーおよび患者の個人情報保護(匿名化管理)も厳格に行わなければなりません。この複雑な情報管理体制の構築は、GCTP運用の大きな山場の一つです。
無菌操作を行う製造要員に対する教育訓練と適格性評価
最終滅菌ができない再生医療等製品において、品質の最後の砦となるのは「人」です。無菌操作を行う製造要員の技術レベルが、そのまま製品の無菌性保証に直結します。
そのため、GCTP準拠の現場では、座学だけでなく、培地充填試験(メディアフィルテスト)などの実技を通じた教育訓練と適格性評価が必須です。定期的に手技の確認を行い、認定された者だけが重要な作業に従事できるような資格制度を社内で設けることが推奨されます。人のスキルへの依存度が高い分、教育訓練プログラムの質が問われます。
生物由来原料基準への適合とウイルス安全性確保
原材料となる細胞や組織、そして培地に使用される血清などは、「生物由来原料基準」に適合していなければなりません。これは、未知のウイルスやプリオンなどの感染性因子が製品に混入するのを防ぐための基準です。
実務では、原材料サプライヤーからの試験成績書(COA)の確認に加え、必要に応じて自社でのウイルス否定試験を実施します。また、マスターセルバンク(MCB)やワーキングセルバンク(WCB)を構築する際にも、徹底した特性解析と安全性試験が求められます。原料調達段階からのリスク管理が、GCTP運用の基礎となります。
品質リスクマネジメント(QRM)に基づく管理基準の設定
GCTP運用において、すべてのリスクをゼロにすることは現実的に不可能です。そこで重要になるのが、品質リスクマネジメント(QRM)の考え方です。リスクの発生確率と重大性を評価し、許容可能なレベルまで低減させるための管理策を講じます。
例えば、閉鎖系システムを導入して汚染リスクを下げることで、環境モニタリングの頻度を調整するといった判断もQRMに基づきます。科学的な知見に基づいてリスクを評価し、メリハリのある管理基準を設定することで、効率的かつ効果的な品質保証体制を構築することができます。
まとめ

本記事では、再生医療特有の品質基準であるGCTPについて、GMPとの違いや実務上のポイントを中心に解説してきました。要点を振り返りましょう。
- GMPとGCTPの違い:GCTPは再生医療等製品の「不均一性」や「無菌化の困難さ」に対応した、柔軟かつ厳格な基準です。
- 特性への対応:個体差への対応、無菌操作の徹底、有効期間の短さを考慮した出荷判定が求められます。
- 実務の要点:交差汚染防止、トレーサビリティ確保、教育訓練、生物由来原料の管理、そしてリスクマネジメントが運用の鍵です。
再生医療分野での品質管理は、単に法令を守るだけでなく、患者様の命に直結する「細胞」という生命の萌芽を預かる責任ある業務です。GCTPの精神を深く理解し、自社の製品特性に合わせた最適な品質管理システムを構築・維持していくことが、再生医療の発展と信頼獲得につながるでしょう。
GMPとGCTP:再生医療特有の品質基準についてよくある質問
再生医療の現場担当者から頻繁に寄せられる、GMPとGCTPに関する疑問をQ&A形式でまとめました。実務の参考としてご活用ください。
- Q1. GCTPはGMPと何が一番違いますか?
- A1. 最大の違いは、原材料(細胞)の個体差や不均一性を前提としている点です。そのため、画一的な管理だけでなく、ベリフィケーション(検証)やリスクベースのアプローチがより重視されます。
- Q2. 治験薬GMPとGCTPの関係はどうなっていますか?
- A2. 治験段階の再生医療等製品には「治験薬GMP」が適用されますが、その考え方はGCTPと整合しています。開発段階からGCTPを見据えた管理体制を構築することが、スムーズな承認申請につながります。
- Q3. 再生医療等製品の記録保存期間はどのくらいですか?
- A3. 原則として、製品の有効期間に10年を加算した期間、または製造日から10年のいずれか長い期間です。ただし、特定生物由来製品に該当する場合は、使用後30年間の保存が求められることがあります。
- Q4. ベリフィケーションとは何ですか?バリデーションとどう違いますか?
- A4. バリデーションは「プロセス全体が恒常的に品質を保証できるか」を確認するものですが、ベリフィケーションは「個々の製造結果が基準を満たしているか」を検証するものです。個体差が大きい再生医療では、この両輪での管理が重要です。
- Q5. 構造設備の要件で最も気をつけるべきことは?
- A5. 「交差汚染の防止」です。特に複数の患者や異なる製品を扱う場合、空調システムによる差圧管理や、動線の分離、閉鎖系機器の導入など、ハードウェア面での物理的な封じ込め対策が極めて重要になります。