PMDAの実地調査や内部監査を控え、準備に追われているご担当者様も多いことでしょう。再生医療等製品の製造管理および品質管理において、GCTP(Good Gene, Cellular, and Tissue-based Products Manufacturing Practice)への適合は絶対条件ですが、その要求事項は多岐にわたり、準備の抜け漏れを防ぐのは容易ではありません。
この記事では、再生医療の現場で品質保証や製造に携わる皆様に向けて、監査対応の実務で即座に活用できる「GMP監査対応チェックリスト」を詳細に解説いたします。品質システムから構造設備、製造管理、バリデーションに至るまで、監査官の視点を踏まえた重要ポイントを網羅しました。ぜひ、貴社の監査準備にお役立てください。
再生医療GMP(GCTP)監査対応に必須となるチェックリストの構成要素
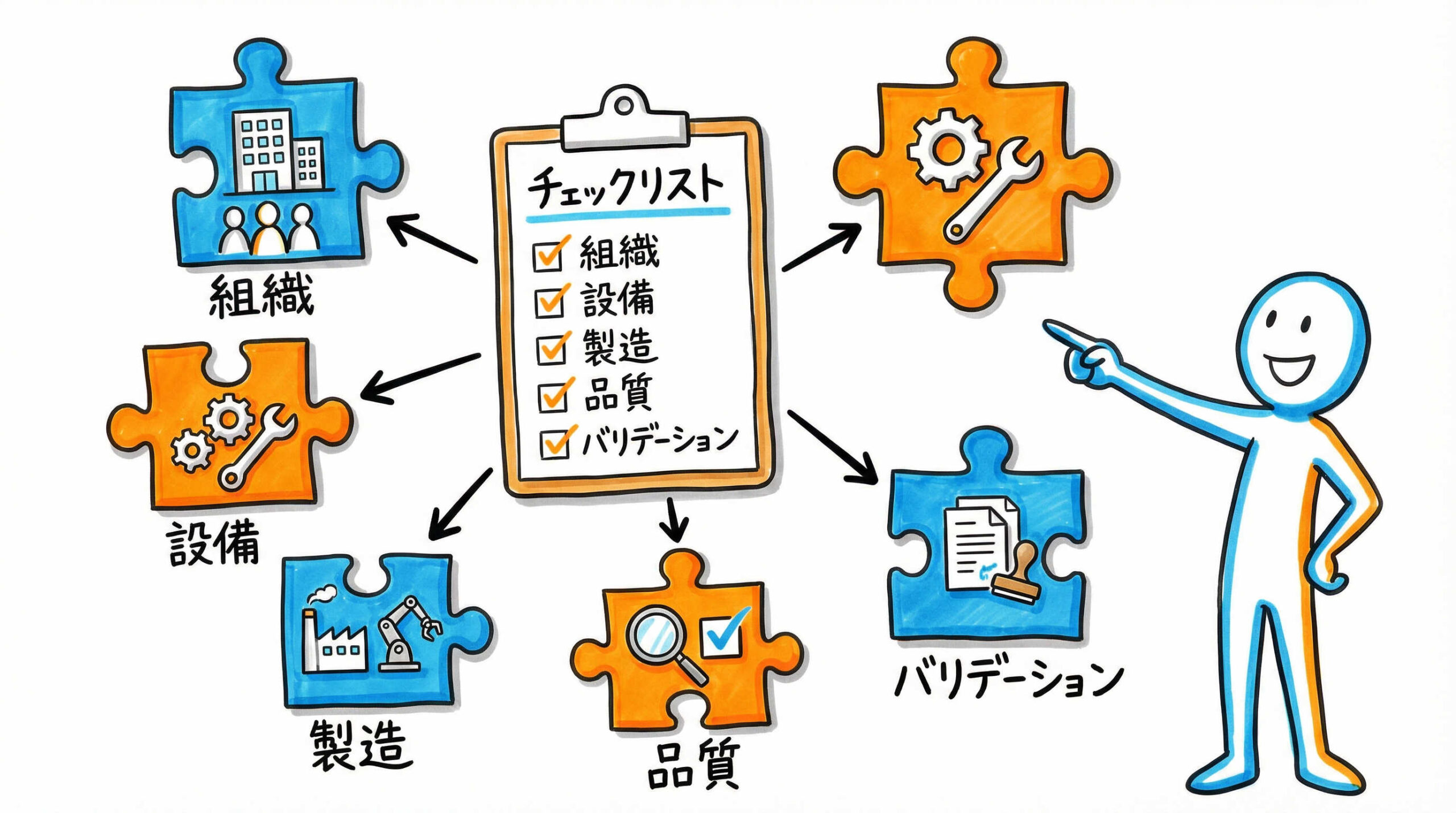
再生医療等製品の監査において、指摘事項を回避し、高い品質保証体制を示すためには、GCTP省令に基づいた体系的な準備が不可欠です。まずは、監査対応チェックリストの骨格となる5つの主要な構成要素について、その重要性と全体像を確認していきましょう。これらを網羅的に押さえることが、監査成功への第一歩となります。
品質システム(医薬品品質システム)に関連する項目
品質システム(PQS)は、組織全体で品質を保証するための基盤となる仕組みです。監査では、単に手順書が存在するかだけでなく、そのシステムが有効に機能しているかが厳しく問われます。
- 経営陣の関与: 品質に対する責任とコミットメント
- 品質目標: 具体的な目標設定と達成状況のレビュー
- 継続的改善: 内部監査やマネジメントレビューを通じた改善サイクル
文書体系の整合性や、組織としての品質文化が根付いているかどうかが、チェックリストの最優先項目となるでしょう。
構造設備および製造環境に関連する項目
細胞という「生き物」を扱う再生医療において、構造設備や製造環境の管理は製品の品質に直結する極めて重要な要素です。無菌性を担保するためのハードウェア面での対策が適切かどうかが焦点となります。
- 清浄度管理: 区域ごとの清浄度区分と管理基準の妥当性
- 空調システム: 差圧管理や気流制御による汚染防止
- 動線管理: 人と物の動線分離による交叉汚染リスクの低減
これらの設備が設計通りに稼働し、維持管理されていることをデータで示す準備が必要です。
製造管理および衛生管理に関連する項目
製造管理および衛生管理は、日々のオペレーションがGCTP省令に則って行われているかを確認する項目です。特に無菌操作が中心となる再生医療では、職員の衛生管理と手技の確実性が重視されます。
- 無菌操作: 手順の遵守とメディアフィルテストによる検証
- 職員の衛生: 健康管理、更衣手順、手洗いの徹底
- 記録の管理: 製造指図書に基づく正確かつリアルタイムな記録(ALCOA+原則)
ヒューマンエラーを防止する仕組みと、逸脱が生じた際の対応力が問われるでしょう。
品質管理(試験検査)に関連する項目
品質管理部門には、製品が出荷に値する品質を備えていることを科学的に証明する役割があります。試験検査の信頼性を確保するための管理体制がチェックリストの要となります。
- 検体管理: サンプリングから保管、廃棄までのトレーサビリティ
- 試験機器: 定期的な校正(キャリブレーション)と保守点検
- OOS対応: 規格外試験結果が出た際の調査手順と原因究明
試験結果の生データ(Raw Data)の管理も含め、データの完全性(Data Integrity)が厳しくチェックされます。
バリデーションおよび文書管理に関連する項目
バリデーションは、製造プロセスや試験方法が期待される結果を恒常的に生み出すことを検証し、文書化する活動です。再生医療等製品は個体差の影響を受けやすいため、プロセスの妥当性評価は特に慎重に行う必要があります。
- バリデーション計画: マスタープラン(VMP)に基づく計画的な実施
- 変更管理: 変更時の再バリデーションの要否判断
- 文書管理: 計画書、実施記録、報告書の承認フローと保管
文書類が最新の状態に保たれ、変更の履歴が追跡可能であることが求められます。
1. 品質システム・組織管理に関する詳細チェック項目
ここからは、より実践的なチェック項目に入っていきましょう。まずは組織の要となる「品質システム・組織管理」についてです。監査では、責任体制の明確化や、問題発生時の自浄作用が機能しているかが重点的に確認されます。以下の詳細項目を一つひとつチェックし、現状の運用と乖離がないか確認してみてください。
製造管理者および品質保証責任者の責務と権限
製造管理者および品質保証責任者が、法令で定められた業務を適切に遂行できる体制にあるかは、監査の基本中の基本です。
- 任免の記録: 適切な資格要件を満たす者が任命され、文書化されているか
- 権限の明確化: 組織図や業務分掌規程において、責任と権限が明記されているか
- 指揮命令系統: 製造部門と品質保証部門が独立し、牽制機能が働いているか
特に品質保証責任者が出荷可否判定を行う際、製造部門からの不当な圧力を受けない体制が確保されていることが重要です。
職員の教育訓練計画と実効性の評価記録
再生医療等製品の製造には高度な専門性が求められるため、職員への教育訓練は品質確保の生命線と言えます。
- 年間計画の策定: 個々の職員のスキルレベルに応じた計画が立てられているか
- 実施記録: 誰が、いつ、どのような教育を受け、理解度はどうだったか
- 効果確認: 教育後の認定制度や実技評価が適切に行われているか
単に座学を行った記録だけでなく、その教育が実務能力の向上に寄与していることを示す「実効性の評価」まで踏み込んで確認しましょう。
変更管理プロセスの運用状況と承認フロー
製造プロセスや試験方法、設備などに変更が生じる際、品質への影響評価なしに変更が行われることは許されません。
- 変更申請: 変更の理由と内容、品質への影響評価が明確に記載されているか
- 承認フロー: 責任者による事前の承認が得られているか
- 事後確認: 変更後の製品品質に問題がないことの検証(フォローアップ)
些細な変更であっても、正式な手順を経ずに現場判断で行っていないか、現場へのヒアリングも含めて確認することをお勧めします。
逸脱発生時の報告ルートと根本原因分析(RCA)
逸脱は「起こさないこと」も大切ですが、「起きた後にどう対応するか」が監査ではより重要視されます。隠蔽体質がないことを示すためにも、報告ルートの透明性が鍵となります。
- 即時報告: 逸脱発生時に速やかに責任者へ報告される仕組みがあるか
- 影響評価: 製品品質への影響範囲が適切に特定されているか
- 根本原因分析(RCA): 「なぜ」を繰り返し、真の原因(ヒューマンエラーで終わらせない)に到達しているか
RCAの手法(Fishbone図や5 Whysなど)を活用した分析記録を整備しておきましょう。
是正措置・予防措置(CAPA)の進捗管理
逸脱や苦情、自己点検などで特定された問題に対し、再発防止策(CAPA)が確実に実行されているかは、品質システムの健全性を測るバロメーターです。
- 計画の妥当性: 原因を除去するための具体的なアクションプランがあるか
- 進捗管理: 期限内に措置が完了しているか、遅延時の対応はどうか
- 有効性評価: 措置実施後、同様の問題が再発していないかを確認しているか
CAPAが「やりっぱなし」にならず、クローズされるまで追跡管理されていることをリストで確認してください。
品質リスクマネジメント(QRM)の活用状況
品質リスクマネジメント(QRM)は、限られたリソースをリスクの高い領域に集中させるための有用なツールです。監査対応においても、QRMの活用は説得力のある説明材料となります。
- リスク特定: プロセス全体のリスクが洗い出されているか
- リスク評価: 発生頻度、重大性、検出性に基づき評価されているか
- リスク低減: 受容可能なレベルまでリスクを下げる措置が講じられているか
ICH Q9などのガイドラインを参考に、リスクアセスメントシートが適切に作成・更新されているか確認しましょう。
自己点検の実施計画と指摘事項への対応
外部からの監査を待つのではなく、自ら問題点を見つけ改善する「自己点検」の実施状況は、企業の自律性を如実に表します。
- 点検計画: 全てのGMP/GCTP要件を網羅するような計画になっているか
- 指摘事項への対応: 発見された不備に対してCAPAが発行されているか
- 経営陣への報告: 結果がトップマネジメントに報告され、リソース配分等の判断材料にされているか
形式的な点検にならず、厳しい目で内部監査が行われている記録こそが、PMDA監査官への信頼に繋がります。
2. 構造設備・製造環境に関する詳細チェック項目
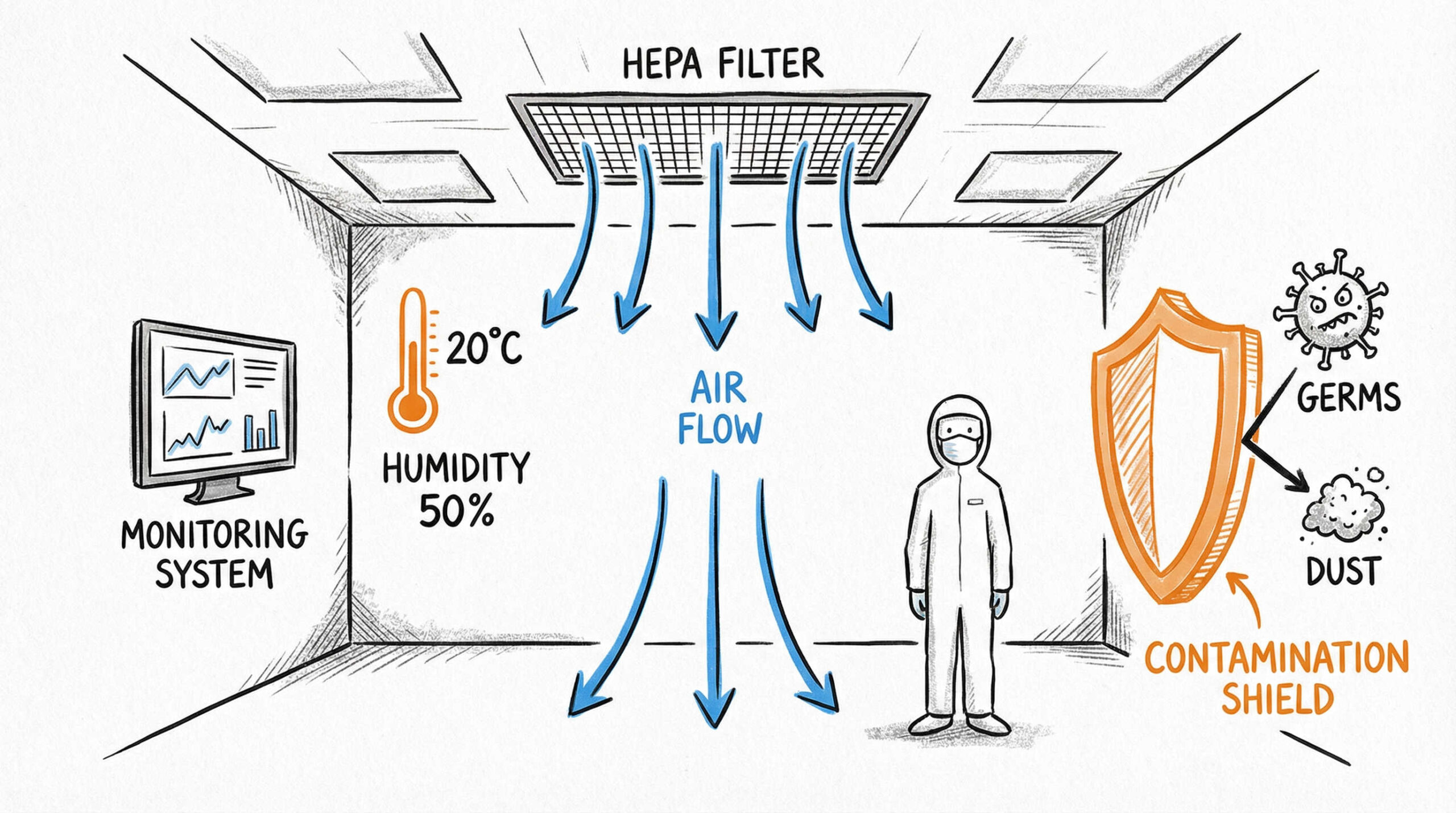
続いて、ハードウェア面である「構造設備・製造環境」のチェックリストです。再生医療等製品の品質リスクの多くは、微生物汚染や交叉汚染に起因します。そのため、設備が清浄に保たれ、その状態がモニタリングされていることを証明するデータは、監査において極めて強力な証拠となります。
細胞調製施設(CPF)の清浄度区分と入退室管理
細胞調製施設(CPF)の清浄度区分(グレードA~D等)が作業内容に応じて適切に設定され、維持されているかは必須チェック項目です。
- 入退室管理: 許可された職員のみが入室できるセキュリティシステム
- 更衣室の運用: 清浄度レベルに応じた更衣手順と動線(一方通行など)
- インターロック: パスボックスやドアのインターロック機能の作動確認
人の出入りは最大の汚染源となり得るため、入退室ログと実際の運用ルールに矛盾がないか、現場で確認することをお勧めします。
空調システム(HVAC)の差圧維持とモニタリング
目に見えない空気の流れを制御し、汚染物質の侵入や拡散を防ぐ空調システム(HVAC)は、CPFの心臓部とも言えます。
- 差圧の維持: 清浄区域から非清浄区域へ空気が流れる陽圧(または封じ込めのための陰圧)設定
- モニタリング: 差圧計の数値が基準値内であることの常時監視と警報システム
- フィルター管理: HEPAフィルターのリークテスト実施記録と交換履歴
差圧の逆転が起きた際の対応手順や復旧記録も、監査で見られるポイントの一つです。
交叉汚染防止のための動線分離(人・物)
複数の患者様の細胞や異なる製品を扱う場合、取り違えや交叉汚染(クロスコンタミネーション)の防止は絶対条件です。
- 動線分離: 原材料搬入、製品搬出、職員の動線が交差しない設計
- 時間的分離: 同一エリアで異なる作業を行う場合の清掃・消毒とインターバル
- 専用化: 可能な限り専用の器具や設備を使用しているか
動線図面と実際の運用を照らし合わせ、物理的または運用面での分離が確実に行われているかチェックしましょう。
安全キャビネットおよびインキュベーターの定期点検
細胞操作の主要な舞台となる安全キャビネット(BSC)やインキュベーターは、日常的な点検が欠かせません。
- 定期点検: 風速、気流、フィルター性能などの定期的な測定(年1回以上)
- 清掃・消毒: 作業前後の清掃手順と使用薬剤の適切性
- 庫内環境: インキュベーターの温度、CO2濃度の記録と校正
特にCO2インキュベーターはカビ等の汚染リスクがあるため、洗浄・滅菌の頻度や手順がSOP通りか確認してください。
製造用水・ガスの品質モニタリング記録
製造に使用する水やガスが汚染されていれば、製品そのものが汚染されます。ユーティリティの品質管理も重要なチェック対象です。
- 製造用水: エンドトキシン、生菌数、導電率、TOCなどの定期測定
- ガス(CO2, N2等): 無菌フィルターの設置と交換管理、純度確認
- トレンド分析: 季節変動や経年変化による水質・ガス品質の推移監視
サンプリングポイントがリスクに基づいて設定されているか(ユースポイントでの採取など)も確認しておきましょう。
環境モニタリング(浮遊菌・落下菌・表面付着菌)の実施
製造環境が「無菌操作にふさわしい状態」であることを証明するのが環境モニタリングです。
- 測定頻度: 作業時(In-operation)と非作業時(At-rest)の適切な設定
- 測定項目: 浮遊菌、落下菌、表面付着菌、浮遊微粒子
- アラート/アクションレベル: 基準値を超えた際の対応手順と逸脱処理
モニタリングポイントが「最悪条件(ワーストケース)」を考慮して設定されているか、その根拠資料(リスクアセスメント)も準備しておくと安心です。
防虫防鼠対策の実施状況とトレンド分析
昆虫やネズミなどの侵入は、GMP施設として致命的な欠陥とみなされます。外部委託している場合でも、管理責任は製造業者にあります。
- トラップ配置: 侵入経路を考慮した適切な配置図
- 点検記録: 定期的な捕獲調査と生息痕跡の確認
- トレンド分析: 季節ごとの虫の発生傾向の把握と対策強化
捕獲数が増加傾向にある場合、建屋の隙間埋めや照明の変更など、具体的な是正措置が取られているか確認しましょう。
3. 製造管理・衛生管理に関する詳細チェック項目
ここでは、実際の製造現場での動きに焦点を当てた「製造管理・衛生管理」のチェック項目を確認します。再生医療等製品は「プロセスが製品(The process is the product)」と言われるほど、製造工程の管理が品質に直結します。手順の遵守と記録の正確性が何より求められる領域です。
原材料・資材の受入試験と合格証の管理
良質な製品は良質な原材料から生まれます。受入時のチェック体制に不備がないか見直しましょう。
- 外観確認: 梱包の破損や汚れがないか
- 試験検査: 規格に適合しているかの確認(試験成績書の確認を含む)
- 合格証: 合格した資材のみが使用可能となるような表示(ラベル等)と保管区分
不合格品が誤って製造ラインに投入されないよう、システム上または物理的に隔離されていることを確実にしてください。
ドナーおよび細胞のトレーサビリティ確保
どのドナーの細胞がどの製品に使われ、どの患者様に投与されたか。この双方向の追跡可能性(トレーサビリティ)は、再生医療等製品の安全性確保における最重要事項の一つです。
- 個体識別コード: 原材料から最終製品まで一貫した識別管理
- 記録の紐付け: 製造記録、試験記録、出荷記録のリンク
- 情報の保存: 法令で定められた期間(製品によっては数十年)、記録が検索可能な状態で保存されているか
万が一の感染症発生時などに、迅速に遡及調査ができる体制を整えておく必要があります。
無菌操作手順の遵守とメディアフィルテストの結果
最終滅菌ができない再生医療等製品において、無菌操作技術は品質の砦です。
- SOP遵守: 手指消毒、物品の搬入出、操作手順が標準作業手順書通りか
- メディアフィルテスト: 定期的(通常半年に1回以上)な培地充填試験によるプロセスシミュレーション
- 要員認定: メディアフィルに合格した職員のみが製造に従事しているか
メディアフィルテストで不合格が出た場合の再教育や再試験の手順、原因究明の記録も重要な監査対象です。
製造指図書に基づく製造記録の正確な記載
「記録にないことは実施していないとみなす」のがGMP監査の鉄則です。製造指図書と製造記録書は、製造の正当性を証明する唯一の証拠です。
- 指図内容: 製造手順、重要工程パラメータ、注意事項が明確か
- 記録の記載: ダブルチェック、実施者の署名、実施日時の正確な記載
- 訂正方法: 訂正印や署名、訂正理由の記載など、訂正ルール(見え消し等)の遵守
記録の不備(空欄、鉛筆書き、修正液の使用など)は典型的な指摘事項ですので、徹底的なレビューを行いましょう。
ロット構成の妥当性と識別管理
ロット(バッチ)の定義と管理は、製品の均質性を保証し、異常発生時の範囲を特定するために不可欠です。
- ロット構成: 1回の製造サイクルや培養単位など、ロットの定義が明確か
- 識別表示: 製造中の容器や保管庫に、品名・ロット番号・工程状態(製造中、試験中など)が表示されているか
- 混同防止: 類似名称の製品や異なるロットが近接して置かれていないか
特に培養期間が長い製品では、工程ごとの識別管理が煩雑になりがちですので、間違いが起きない仕組み作りが重要です。
職員の更衣手順および健康状態の確認
職員自身が最大の汚染源とならないよう、更衣と健康状態の管理は厳格に行う必要があります。
- 更衣手順: 鏡による確認や、肌の露出を最小限にする手順が守られているか
- 健康状態: 発熱、下痢、皮膚疾患などの症状がある職員の作業制限ルール
- 健康診断: 定期的な健康診断と、必要に応じたワクチン接種等の感染症対策
体調不良者が無理をして作業に入らないよう、申告しやすい職場環境やバックアップ体制があるかどうかも、間接的に品質に関わってきます。
設備の清掃・消毒手順と実施記録(ログブック)
製造終了後の清掃・消毒は、次の製造への準備であり、交叉汚染防止の要です。
- SOPの具体性: 消毒剤の種類、濃度、接触時間、拭き取り方法が具体的に定められているか
- 実施記録(ログブック): 誰が、いつ、どの設備を清掃したかの記録
- 消毒剤の管理: 消毒剤の有効期限管理と、耐性菌出現防止のためのローテーション
ログブックは監査官が必ずと言っていいほど確認する資料です。記載漏れや不整合がないか、日常的にQAが確認する体制にしましょう。
4. 品質管理・バリデーションに関する詳細チェック項目

製品の品質を科学的に担保する「品質管理」と、プロセスの妥当性を証明する「バリデーション」。これらは監査において論理的な整合性が厳しく問われる分野です。データの信頼性(Data Integrity)を意識しながら、以下の項目をチェックしていきましょう。
試験検査設備の校正(キャリブレーション)記録
試験結果の信頼性は、使用する機器が正しく計測できているかどうかに依存します。
- 校正計画: 全ての重要機器について、校正周期と許容範囲が設定されているか
- トレーサビリティ: 国家標準などの標準器にトレーサブルな校正が行われているか
- 有効期限表示: 機器本体に校正済証(有効期限付き)が貼付されているか
校正の結果、基準を外れていた場合、過去に遡ってその機器で測定した製品への影響評価を行っているかも確認が必要です。
規格外試験結果(OOS)発生時の調査手順
規格外試験結果(OOS)が発生した際、安易に再試験を行って合格にしていないでしょうか。これは重大な指摘事項になり得ます。
- 初期調査: 実験ミスや機器トラブルなどの明白な原因(Assignable Cause)の有無
- 拡大調査: 製造工程の調査や過去のデータの照査
- 再試験の正当性: 科学的な根拠に基づいた再試験プロトコルの作成と承認
OOSは「品質改善のチャンス」と捉え、根本原因を究明し、再発防止につなげるプロセスが回っていることが重要です。
検体の採取方法と保管管理(参考品・保存品)
検体(サンプル)の取り扱いは、試験結果の正当性を左右します。また、参考品・保存品の管理は将来の検証のために必須です。
- サンプリング: 代表性を確保した採取方法と汚染防止策
- 保管条件: 製品の安定性を損なわない温度・湿度での保管とモニタリング
- 参考品・保存品: 法令で定められた期間・数量の保管と、定期的な目視確認
検体の取り違え防止策や、保管庫のセキュリティ(鍵の管理など)も合わせてチェックしましょう。
バリデーションマスタープラン(VMP)の整備
バリデーション活動の全体像を示すマスタープラン(VMP)は、監査官に貴社のバリデーション方針を説明する地図のようなものです。
- 対象範囲: 施設、設備、プロセス、洗浄、試験法など、対象となるバリデーションの範囲
- スケジュール: 年間の実施計画とリソース配分
- 責任体制: バリデーション委員会や責任者の明確化
VMPと個別の実施計画書、報告書の整合性が取れているか、日付や版数を含めて確認してください。
プロセスバリデーションおよび無菌性保証の検証
再生医療等製品では、製品そのものでの全数検査が難しいため、プロセスのバリデーション(PV)による無菌性保証が極めて重要です。
- 重要工程パラメータ(CPP): 品質に影響を与えるパラメータの特定と管理幅の設定
- 3ロット検証: 連続する3ロット以上での恒常性の確認(またはコンカレントバリデーションの適用正当性)
- 無菌性保証: 無菌操作法バリデーション(メディアフィル)との連携
製品の特性上、PVの実施が困難な場合でも、それに代わる検証アプローチ(ベリフィケーション等)の妥当性を論理的に説明できるようにしましょう。
分析法バリデーションの実施状況
試験方法が目的に適しており、信頼できる結果を出すことを証明するのが分析法バリデーションです。
- 特性評価: 特異性、直線性、範囲、真度、精度、検出限界、定量限界などの評価
- 公定法との比較: 日本薬局方などの公定法以外の試験法を採用する場合の同等性評価
- 技術移転: 開発部門からQC部門への試験法移管時の比較試験
特に、キットを用いた試験や独自の試験法を採用している場合は、その妥当性を示すデータの提示が求められます。
輸送バリデーション(輸送中の温度管理等)
細胞は温度変化や振動に敏感です。医療機関へ届くまでの品質を保証するため、輸送バリデーションも見逃せません。
- 容器の性能: 温度保持能力(バリデーション済み輸送容器の使用)
- 輸送ルート: 想定される輸送時間や外気温の影響評価
- 温度ロガー: 実際の輸送時の温度記録と逸脱時の対応
輸送業者への委託契約内容(責任分界点や品質取り決め)も、監査で確認されるポイントの一つです。
再生医療等製品特有のGCTP監査で重視されるポイント

再生医療等製品(GCTP)には、一般的な医薬品GMPとは異なる特有の要求事項があります。これらは製品の安全性、特にウイルス等の感染症リスク管理に直結するため、監査において最優先で確認される項目です。ここをクリアしなければ、再生医療等製品としての製造は認められないと言っても過言ではありません。
生物由来原料基準への適合性確認
再生医療等製品の製造に使用する原材料(培地、血清、酵素、成長因子など)は、「生物由来原料基準」への適合が必須です。
- 原産国確認: 反芻動物由来原料の場合、BSE/TSEリスクのない原産国であるか
- ウイルス不活化: 製造工程でのウイルス除去・不活化処理の記録
- 適格性確認: サプライヤーからの証明書(CoA)だけでなく、自社での評価記録
「研究用試薬(RUO)」を使用せざるを得ない場合でも、リスク評価を行い、必要な安全性試験を実施していることの根拠資料を揃えておく必要があります。
細胞バンク(マスター・ワーキング)の管理体制
細胞バンク(マスターセルバンク:MCB、ワーキングセルバンク:WCB)を構築している場合、その管理は製品の恒常性を左右します。
- 特性解析: 遺伝的安定性、増殖能、生存率などの評価データ
- 保存管理: 液体窒素タンク等の温度管理、バックアップ保管(分散保管)
- 更新手順: 新しいバンクを作製する際の手順と評価基準
バンクの履歴管理(いつ、誰が、どの株から作製したか)は、将来にわたって参照される重要な記録となります。
マイコプラズマ否定試験およびウイルス安全性試験
細胞製品におけるマイコプラズマやウイルスの混入は、患者様の命に関わる重大なリスクです。
- 否定試験: 培養工程の適切な段階(中間製品、最終製品など)での実施
- 試験感度: 核酸増幅法(NAT)などを採用する場合のバリデーション(検出限界の証明)
- 外来性ウイルス: 原材料や細胞株に由来するウイルスリスクの網羅的な否定
迅速法を採用する場合は、公定法との同等性あるいは優越性を示すデータ整理が不可欠です。
治験薬GMPから商用GMPへの移行時の同等性評価
治験段階から商用製造へ移行する際、製造スケールの拡大や設備の変更が行われることがよくあります。この時の「同等性(Comparability)」の評価は、審査の大きな山場です。
- 品質特性の比較: 治験製品と商用製品の品質属性(CQA)の比較データ
- 変更管理: 製造プロセス変更の前後でのバリデーションデータの蓄積
- リスク評価: 変更が安全性や有効性に与える影響の科学的考察
「治験の時と同じ品質のものが作れている」ことを、客観的なデータで語れるように準備しましょう。
PMDA実地調査・内部監査を成功させる事前準備の進め方
チェックリストによる現状確認ができたら、次は監査当日に向けた具体的な準備に入ります。PMDAの実地調査や内部監査をスムーズに進め、信頼を勝ち取るためには、事前のシミュレーションと体制づくりが鍵を握ります。ここでは、成功率を高めるための準備の進め方をご紹介します。
模擬監査による指摘事項の洗い出しとランク付け
本番さながらの模擬監査(モック監査)は、欠かせないプロセスです。外部コンサルタントや、他部署の経験豊富な社員を監査員役として招くと効果的です。
- 指摘事項の洗い出し: 客観的な視点で不備を見つけ出し、リスト化する
- ランク付け: 重篤度(クリティカル、メジャー、マイナー)に応じて優先順位をつける
- 回答訓練: 質問に対する的確な回答(余計なことを言わない、事実のみを述べる)の練習
模擬監査で見つかった不備に対し、本番までに是正が間に合わない場合でも、「課題として認識し、計画的に対応中である」と説明できる準備をしておくことが重要です。
バックヤード支援体制と資料提示のシミュレーション
監査当日は、監査対応ルーム(フロント)と資料準備ルーム(バックヤード)の連携が勝負を分けます。
- 役割分担: 記録係、資料検索係、ランナー(資料を運ぶ係)、司令塔を明確にする
- 資料リストの整備: どの資料がどこにあるか、即座に検索できるインデックス作り
- リハーサル: 監査官からの資料要求から提示までのタイムラインを測定し、短縮を図る
スムーズな資料提示は、「普段から整理整頓されており、管理が行き届いている」というポジティブな印象を与えます。
既往の指摘事項に対する改善状況の再確認
過去の監査(PMDA調査や内部監査、ベンダー監査など)で指摘された事項が、今回も改善されていない場合、監査官の心証は極めて悪くなります。
- CAPAの完了確認: 過去の指摘に対する是正措置が完了し、効果が維持されているか
- 記録の再確認: 改善後の運用記録が確実に残されているか
- 説明ロジック: 万が一改善が遅れている場合、正当な理由と進捗状況を説明できるか
「同じ指摘を二度受けない」ことは、監査対応の鉄則です。既往の指摘事項リストを再度見直しましょう。
監査当日のSOP(標準作業手順書)即時検索体制の構築
監査中、手順の詳細について質問された際、SOP(標準作業手順書)を即座に提示できる体制は必須です。
- 電子化と検索: 電子ファイルでキーワード検索できる環境、または紙媒体の分かりやすいファイリング
- 最新版管理: 提示したSOPが最新版であることを確実に保証する
- 現場との整合性: 現場に備え付けのSOPと、監査室で提示するマスターファイルが一致しているか
「確認して後ほど回答します」が多すぎると、管理能力を疑われかねません。主要なSOPはすぐに取り出せるようにしておきましょう。
まとめ
再生医療GMP(GCTP)監査への対応は、一朝一夕で成し遂げられるものではありません。しかし、今回ご紹介した「品質システム」「構造設備」「製造管理」「品質管理」「バリデーション」という5つの柱に基づき、詳細なチェックリストを活用して一つひとつ確認を進めれば、必ず道は開けます。
特に再生医療等製品は、科学的知見の蓄積とともに規制も進化しています。常に最新のガイドラインにアンテナを張り、自社の品質システムをアップデートし続ける姿勢こそが、監査官からの信頼、ひいては患者様への安全・安心につながります。このチェックリストが、貴社の監査準備の一助となり、無事に適合認定を受けられることを心より応援しております。
GMP監査対応チェックリストについてよくある質問

再生医療の現場担当者からよく寄せられる、GMP監査対応に関する質問をまとめました。